
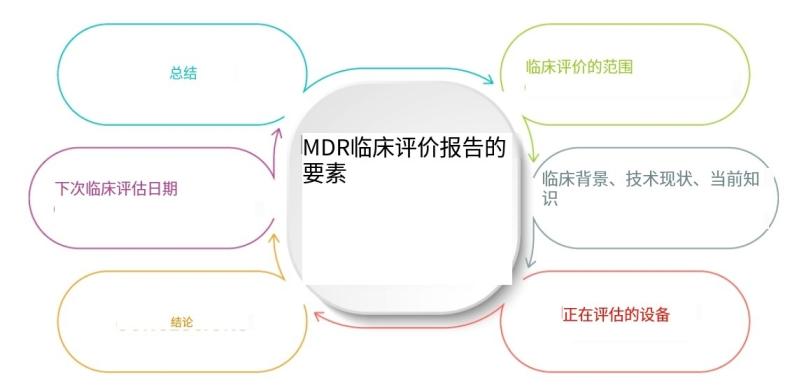
欧盟新的医疗器械法规(MDR)取代了有源植入式医疗器械(AIMD)上的医疗器械(MDD)和90/385 / EEC指令93/42 / EEC。它为医疗设备制造商带来了更严格的要求,并扩大了指定机构的责... 查看详情>
共 127 讨论,7天新增 1 个讨论,30天新增 1 个讨论

 胸有大志
回复了问题
2023-09-17 00:24
胸有大志
回复了问题
2023-09-17 00:20
胸有大志
回复了问题
2023-09-17 00:19
胸有大志
回复了问题
2023-09-17 00:18
胸有大志
回复了问题
2023-09-17 00:17
胸有大志
回复了问题
2023-09-17 00:16
胸有大志
回复了问题
2023-09-17 00:15
胸有大志
回复了问题
2023-09-17 00:14
胸有大志
回复了问题
2023-09-17 00:24
胸有大志
回复了问题
2023-09-17 00:20
胸有大志
回复了问题
2023-09-17 00:19
胸有大志
回复了问题
2023-09-17 00:18
胸有大志
回复了问题
2023-09-17 00:17
胸有大志
回复了问题
2023-09-17 00:16
胸有大志
回复了问题
2023-09-17 00:15
胸有大志
回复了问题
2023-09-17 00:14