
2019年度美国FDA研制环节检查(BIMO)概况
-

-
加菲 这家伙很懒,还没有设置简介
0 人点赞了该文章 · 2830 浏览
FDA近期公布了2019年研制环节的检查工作统计数据。FDA的研制环节检查简称BIMO(Bioresearch Monitoring)检查,包括GCP检查(分别针对研究者、伦理委员会、申办者/监查员/CRO、研究者发起的试验、生物等效性试验)、GLP检查、上市后药品不良事件报告(PADE)检查和风险评估缓解策略(REMS)检查等8个类型,涵盖FDA的全部6个中心,即药品审评中心(CDER)、生物制品中心(CBER)、医疗器械与放射健康中心(CDRH)、食品安全与应用营养中心(CFSAN)、兽药中心(CVM)和烟草产品中心(CTP)。
一、检查数量
2019年,按照被检查机构统计,FDA共发起1402次境内和境外BIMO检查,具体数据见下表。
表1 各中心检查数量统计数据
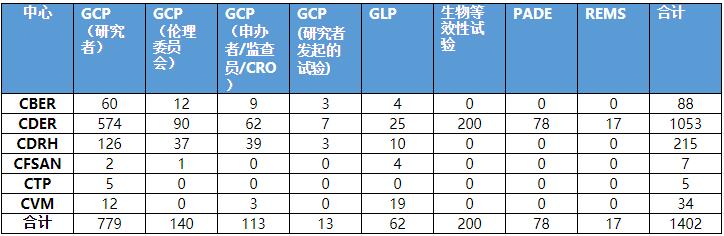
二、检查缺陷
FDA的检查结果按照缺陷的严重程度,分为NAI(No Action Indicated,无需采取后续措施)、VAI(Voluntary Action Indicated,需被检查机构主动整改)和OAI(Official Action Indicated,需要FDA采取监管措施)。
1. GCP检查(研究者)
2019年,FDA共发起779次针对研究者的检查,其中境内检查600次,境外检查179次。境内检查中,NAI为74%,VAI为25%,OAI为1%。境外检查中,NAI为82%,VAI为18%,无OAI。
常见的检查缺陷包括:研究者未遵守试验方案、方案偏离未记录;未遵守研究者声明;原始病历不完整、试验记录不完整;试验用产品的管理记录不完整;受试者保护不足、知情同意不符合法规要求;不良事件未报告或未记录;伦理委员会不符合法规要求等。
2. GCP检查(伦理委员会)
2019年FDA开展了140次伦理委员会的境内检查,无境外检查。其中,NAI为79%(111次),VAI为20%(28次),OAI为1%(1次)。
常见的缺陷包括:伦理审查会议记录不完整、会议表决人数不符合要求;伦理委员会人员组成不合理;伦理的初始审查和跟踪审查不完善;伦理委员会没有制定暂停或终止试验的报告的SOP;伦理委员会记录不完善,试验方案及相关文件未按规定保存。
3. GCP检查(申办者/监查员/CRO)
113次针对申办者/监查员/CRO的检查中,境内检查108次,境外检查5次。境内检查中,NAI为73%,VAI为23%,OAI为4%。境外检查中,NAI为80%,VAI为20%,无OAI。
常见的缺陷包括:申办者未能选择合格的研究者/监查员,未对临床试验进行充分监查,未能确保试验过程遵守试验方案,未按照法规要求保存试验资料,试验用产品的管理记录不完整,对依从性不佳的研究者未及时纠正或终止其资格。
4. GCP检查(研究者发起的试验)
13次针对研究者发起的试验的检查均为境内检查,其中NAI为54%(7次),VAI为46%(6次)。常见的缺陷包括:未提交新药临床试验(IND)申请;未能确保对临床试验进行充分监查;未遵守试验方案;未遵守研究者声明;原始病历不完整、试验记录不完整;试验用产品的管理记录不完整;受试者保护不足、知情同意不符合法规要求;伦理委员会不符合法规要求等。
5. GLP检查
2019年开展了62次GLP检查,其中境内检查54次,境外检查8次。境内检查中,NAI为46%,VAI为33%,OAI为21%。境外检查中,NAI为50%,VAI为50%。
常见的缺陷包括:供试品、试剂等标签信息不完整;专题负责人未及时归档、未记录试验中的意外情况,未确保数据得到准确的记录和核对;SOP不完善;未遵守试验方案;最终报告中未对影响数据质量和可靠性的情况进行讨论。
6. 生物等效性试验(BE)检查
2019年FDA进行了200次生物等效性试验检查,其中境内检查90次,境外检查110次。境内检查中,NAI为83%,VAI为16%,OAI为1%。境外检查中,NAI为85%,VAI为10%,OAI为5%。
常见的缺陷包括:方法学验证不规范,留样不符合要求,未能保持试验的盲态,以及试验记录不完整。
7. 上市后药品不良事件报告(PADE)检查
78次PADE检查中,境内检查69次,境外检查9次。境内检查中,NAI为67%,VAI为33%。境外检查中,NAI为22%,VAI为78%。
常见的缺陷包括:未制定与药品上市后不良事件的监测、接收、评价与报告等相关的SOP;未在15日内提交安全性报告;未及时提交年度定期安全性报告;未及时提交每季度的定期安全性报告;资料保存不完整。
8. 风险评估缓解策略(REMS)检查
17次REMS检查均为境内检查,其中NAI为88%(15次),VAI为12%(2次)。常见缺陷包括:未遵守REMS的用药安全保障措施(ETASU,Elements To Assure Safe Use);未遵守REMS用药指南;未在15日内提交安全性报告;未遵守REMS执行计划。
来源:
全部 0条评论