
知识分享|灭菌制剂的无菌检查
-

-
加菲 这家伙很懒,还没有设置简介
0 人点赞了该文章 · 7852 浏览
一、无菌检查概念及意义
1. 无菌检查的概念及范围
无菌检查法是指检查无菌或灭菌制品、敷料、缝合线、无菌器具及适用于药典要求无菌检查的其他品种是否无菌的一种方法。也就是说,凡直接进入人体血液循环系统、肌肉、皮下组织或接触创伤、溃痛等部位而发生作用的制品或要求无菌的材料、灭菌器具等都要进行无菌检查。对医疗及预防用的生物制品,是指制品不得含有杂菌,灭活菌苗、疫苗不得含有活的病菌、病毒的一套无菌检查法。药品、敷料等应按《中国药典》收载的无菌检查法的规定进行,生物制品应按《中国生物制品规程》收载的无菌检查法的规定进行。
需要进行无菌检查的药品、敷料、灭菌器具的范围主要有以下几类:
(1)各种注射剂:用于肌肉、皮下和静脉的各种针剂,包括注射用的无菌水、溶媒和溶剂、输液、注射剂原料等。
(2)眼用及外伤用制剂:用于眼科手术、角膜创伤及一般创伤、愤殇和烧伤等外科用药品制剂。许多国家对这类制剂均规定为无菌,不得染有任何活菌,我国也基本如此。用于注射或深部组织创伤、溃殇、出血的中药制剂则应无菌。
(3)植入剂:即用于包埋于人体内的药物制剂,如不溶于水的激素、避孕药物、免疫药物及抗肿瘤药物等要求无菌的制剂。心脏瓣膜以及固定用金属板和有机器材等。
(4)可吸收的止血剂:如明胶发泡剂、凝血酶等用于止血并可被组织吸收的各种药物制剂。
(5)外科用敷料、器材:如外科手术用脱脂棉、纱布、结扎线、缝合线、可被吸收的羊肠线及一次性注射器与一次性无菌手术刀片、输血(输液)袋、博士伦等。
按无菌检查法规定,上述各类制剂均不得检出需气菌、厌气菌及真菌等任何类型的活菌。从微生物类型的角度看,即不得检出细菌、放线菌、酵母菌及霉菌等活菌。
无菌检查的结果为无菌时,在一定意义上讲,它要受抽验样本数量的限制,同时也要受灭菌工艺的限制,对最终灭菌品达到10-6的微生物存活概率,就认为灭菌的注射制品合格。所以并非绝对无菌,这个结果也是相对意义的。
2. 无菌检查的意义
1972 年英国普拉姆斯总医院发生了因输注染菌的葡萄糖导致6 个患者死亡。1970年7 月至1叨1 年3 月,美国的25 个医院中由于输注污染的葡萄糖致使378 个患者得了败血症, 40 人死亡。我国也有类似情况发生。1师1 ~ 1993 年人血白蛋白污染,输注后发生46 例感染,死亡8 例。无论哪种制品污染了微生物,对患者的安全与危害是可想而知的。因此人们较早地认识到应对此类制剂进行无菌检查。无菌检查是药品微生物检验的最早的要求,在20 世纪初就列人必检项目。现今世界各国药典均对无菌检查范围、内容、方法以至抽样都有明文规定,用以保证无菌或灭菌制剂等的用药安全。由于无菌或灭菌制剂在医疗、防疫中应用广泛,所以对规定灭菌或无菌制剂进行无菌检查,在保证人民用药安全方面有着十分重要的意义。
二、无菌检查抽样
1. 抽样的意义
无菌检查的抽样,是从一批产品中抽取一定数量单位的样品,作为检验样本的方法。
各种要求无菌的药品在出厂前,应通过无菌检查,以保证产品的质量。但由于每批产品的数量众多,目前由于现有知识及检测手段的限制,提供不了非破坏性确认批量产品中每一个最终制品微生物质量的方法。而目前检查的方法又是破坏性的,不可能对所有产品单位进行检查。因此,通常只能从每批产品中随机抽取一定数量的单位产品,作为样本检验,以此结果来判断整批产品(总体)的质量。
随机抽样,不仅用于药品的无菌检查、药品微生物限度检验,药品的其他质量检查,除特殊情况之外,一般都采用随机抽样方法。
随机抽样中,对分批应特别注意。对无菌检查而言,一个批量应以同一灭菌器的产品为一批:无菌制剂应以无菌灌装相同的最终容器,在连续不间断生产,以不超过24 h的时间周期内的产品为一批;在连续生产过程中产品分别连续灭菌,如γγ-射线灭菌应以不超过24 h 的总产量为一批;不同机器生产的,以各机的产品分批;不同班组生产的,应按班组分批。这样分批的意义是使各批号的产品具有均匀性,随机抽样时则有代表性。此外,尚有其他管理方面的指导意义。
以同一灭菌器中的产品分为一批时,应由不同部位抽取单位产品组成样本。连续生产过程中,应有不同时间抽样组成样本。
2. 抽样量
抽样方法及抽样量是一个相当复杂的问题,目前各国对于无菌检查的药品,有关抽样方法和抽样量的规定都不统一。
抽样方法基本上可分三种类型:
(1)百分数抽样法:本法抽样是根据统计学概率的估算,确定随机抽样的数量。如WHO 于1叨5 年建议的原则,英国药典1998 版的抽样量(表1 - 1 )按各批单位产品的数量确定每批的抽样量,每批抽样量为批量的5% 左右,高者不超过20% ,低者可以在2% 以下。

(2)固定抽样法:每批产品皆抽取固定量的代表样品,而不论每批产品量的多少。
(3 )综合抽样法:很多国家规定的抽样法,常用上述两种综合形式的方法,即固定抽样与按百分数抽样的综合法。固定抽样法多在每批产品众多时采用。
美国药典23 版的无菌试验抽样量(表1 - 2)与前几版不间,取消了抽样方法的详细规定和表格,只规定为每种培养基需用的容器数。如容器药液装量在100 ml 以上,每种培养基需接种来自10 个容器的规定药液量,这是固定抽样量法。而USP 24 版(2000年)则采用了BP 1998 的抽样方法,最多抽20 个容器。

中国药典2015年版无菌检查法规定的抽样量,
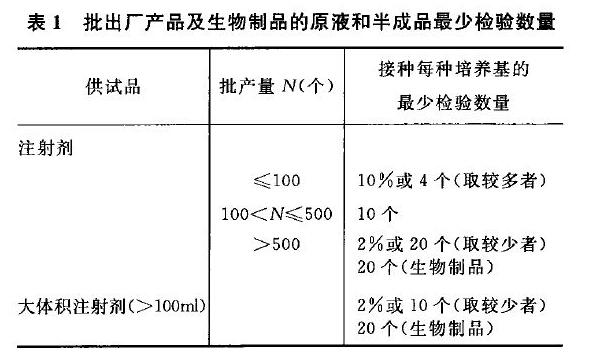
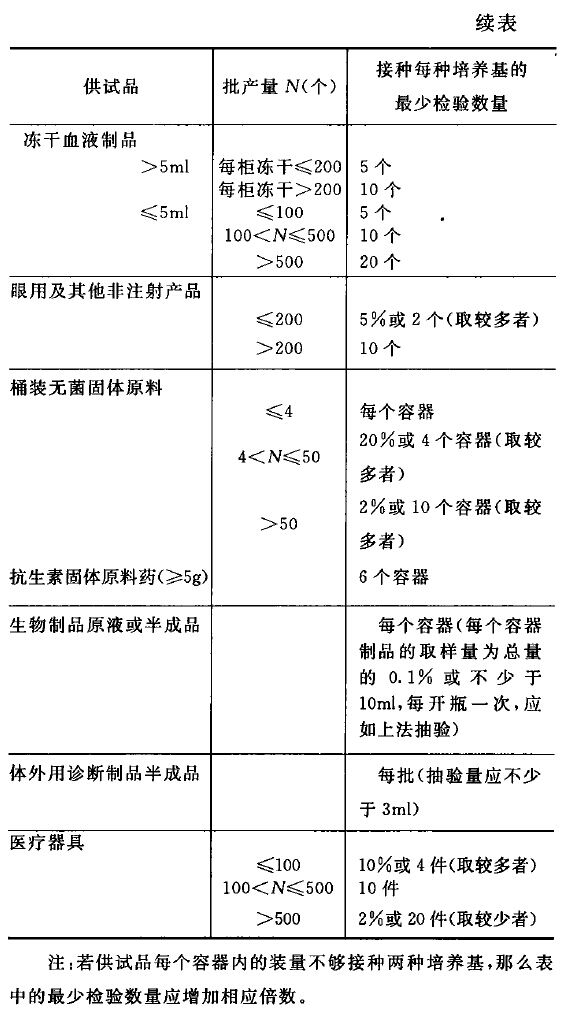

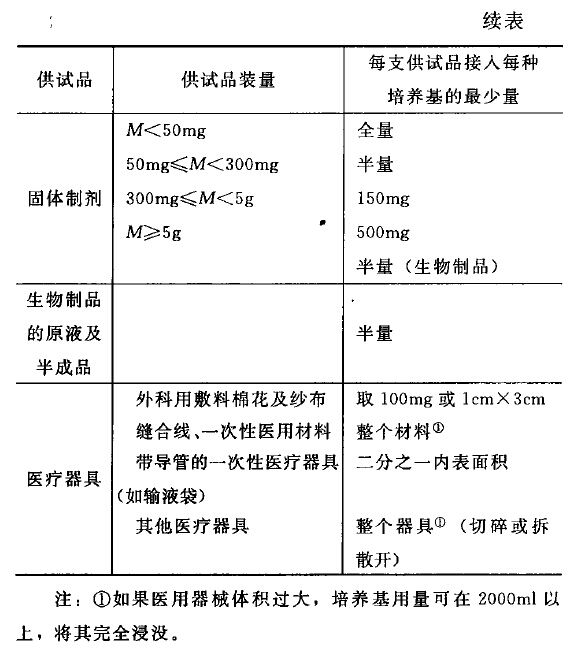
3. 抽样量与合格率
抽样的数量,常受多方面因素的制约,例如检验的目的、要求、代表性及抽样方法,实际工作量及经济损失等因素。总之,力求以最少样本量准确地反映客观情况。
例如因灭菌不完全或无菌灌装过程发生某种意外等,某一批中有10% 的产品受微生物污染,从这个批中取一个成品样本作无菌检查,存在两种可能性,取到无菌样品或取到染菌样品,取到无菌样的概率p 为0.9,而取到染菌样的概率q 为0.1 ,则
(p+q) = (0.9+0.1)1 = 1.0
如从这个批中取2 个成品样本作无菌检查,则存在三种可能: 2 个无菌样本; 2 个染菌样本; 1 个无菌样本和1 个染菌样本。以三种可能组合的概率可按下式计算:
(p + q)2= 1.0 即(p + q)2= p2 + 2pq+ q2 ,将p =0.9 和q =0.1 代人即得
p2=0.81 取得2 个无菌样本的概率
q2 =0.01 取得2 个染菌样本的概率
2pq=0.18 取得1 个无菌样本和1 个染菌样本的概率
因此,当样本数为2 时,这个10% 染菌的批抽不到染菌样本的概率为p2=0 .81 ,抽到染菌样本的概率为0.19。
在无菌检查中,一个染菌批,设样本数为n ,通过元菌检查的概率和这个染菌批被检查出来的概率为
通过无菌检查的概率为pn = ( 1- q )n
检出批染菌的概率为1- pn=1- (1-q)n
q 系批产品的染菌概率,与生产工艺提供无菌保证的能力有关, n 则是药典无菌检查法规定的抽验样本数。不同污染率下所抽样本数通边无菌检查的概率如表1-3:
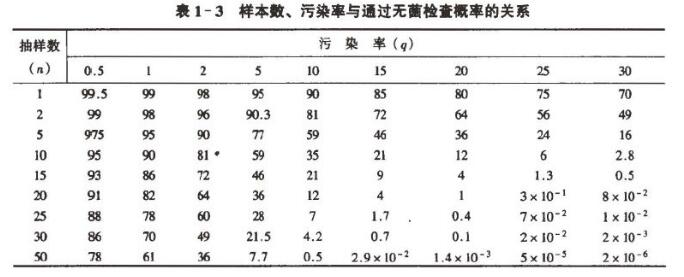
从表1 一3 中的数据及图1 - 1 可以得出:
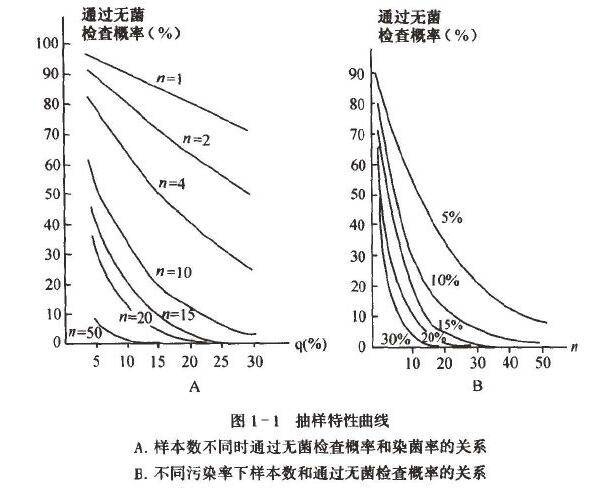
(1) 当批产品污染率越低,即使增大抽样量n ,也很难检出污染的部分。根据元菌检查结果来判断批的无菌,其风险就越大。一个批中有30% 污染率,抽检样本数为2时,通过无菌检查的概率为49% 。当污染率下降至5% 时,若抽检样本数为2 ,则误判合格的概率上升为90.25%0
(2)随着污染率增大,检查不合格的概率上升。
(3)在相同污染率下,增大元菌检查的样本数,可提高无菌检查的可信度。
仪器设备及用具
无菌检查所用的主要仪器设备用具如下:
无菌室为单独隔离的无菌操作间和两个缓冲间组成,结构和材料应符合要求。操作间净化级小于10000 级,局部净化级小于100 级。室温18 - 26℃, 相对温度为40% -60%。
全封闭无菌检测系统、开放式滤器、真空泵、抽滤瓶。
恒温培养箱( 30 ~ 35 ℃)、生化培养箱( 20-25 ℃) 、恒温水浴。
高压蒸汽灭菌器、电热恒温干燥箱(250℃)。
显微镜(1 500x)
玻璃器皿试管、锥形瓶、量筒、培养皿( φ 90 mm)、刻度吸管( 1 、5 、10 ml )、注射器(5 、10 、20 、30 ml )、针头( 9 、11 、12 、16 号) 。
手术慑、剪
无菌衣、裤、帽、口罩、鞋罩。
以上玻璃器皿洗净、晾干,用牛皮纸包扎严密, 121 ℃蒸汽灭菌30 min ,备用。手术慑、剪刀160-180℃干烤2h。
所有灭菌物品存放于第一缓冲间,不应越过2 周即用毕。否则,应重新灭菌。玻璃注射器、针头等小件物品的灭菌,不宜放在带盖的密闭搪瓷盒、铝制饭盒内以高压蒸汽灭菌,因盒内的冷空气不易排尽,而影响灭菌效果。
无菌室应定期用消毒液擦拭并进行净化度检查,如尘埃微粒数及浮游微生物数,应符合规定, 否则不得使用。
灭菌器材、供试品等外包装在缓冲间拆除后,送至净化台上,避免将外包装污染的微生物带进无菌室。
稀释剂 0.9%无菌氯化钠溶液、无菌蒸馏水。USP23 版收载稀释液A (动物组织胃蛋白酶消化物或同类产品1.0% 、蒸馆水加至1000ml,灭菌后pH 值为7 .1 士0.2 ,分装,高压蒸汽灭菌)。A 液用于青霉素、头抱菌素类样品的无菌试验。稀释液 D (按上述稀释液A 中,每1000 ml 溶液加10 ml 聚山梨酸- 80 ,灭菌后pH 值为7.1±0.2,分装,高压蒸汽灭菌)。D 液适用于含卵磷脂、油或用薄膜过滤的样品无菌检查。稀释液K (木瓜酶消化动物组织物5.0 g、 牛肉提取物3.0 g、聚山梨酸- 80 10ml、蒸馏水1000ml,灭菌后pH 值为6.9 土0.2)。K 液适用于在十四皖酸异丙脂溶解的含凡士林的样品。
培养基及培养基灵敏度试验
一、无菌检查用培养基
1. 需气菌、厌氧菌培养基(硫乙醇酸盐培养基)
中国药典1995 年版所规定的硫乙醇酸盐液体培养基(第26 章培养基1) ,与中国生物制品规程的规定一致,与英、美、日等国的药典规定也基本一致。
由于细菌种类繁多,营养要求多样性,因此,要使单一的培养基适应各种细菌的生长要求是困难的。各国在修订药典时,对无菌检查用的培养基也不断改进。
现在采用的硫乙醇酸盐液体培养基,基本上适用于需气菌与厌氧菌的生长要求。该培养基的特点:①膜陈、酵母浸出粉提供氮源以及供合成蛋白质必须的各种氨基酸和B族维生素:②脱氨酸、硫乙醇酸盐、葡萄糖均有降低氧化还原电位的作用,使深部氧化还原电位适合于厌氧菌的生长,同时硫乙醇酸盐有钝化含碑和柔类药物及防腐剂的抑菌作用;③增添刃天青或亚甲蓝作为氧化还原的指示剂。前者有氧时呈红色,后者有氧时呈蓝绿色。由于亚甲蓝的抑菌作用较强,现均用刃天青;④少量琼脂有助厌氧环境的形成,防止因液体对流而迅速产生氧化。
2. 真菌培养基
中国药典1995 版规定的真菌培养基(培养基3),其处方为改良马丁培养基(培养基11 ),与中国药品生物制品规程1995 版收载的真菌培养基是一致的。但与英、美药典的真菌培养基不同。
关于检查真菌用的培养基,各国不尽一致。我国及日本都规定适于霉菌和酵母菌生长的培养基,而英国和美国药典规定的真菌培养基,仅指出用大豆酷蛋白消化物培养基(培养基4),该培养基适用于需气菌和真菌生长。如供试品污染了需气菌,无论接种在需气菌、厌气菌培养基或大豆酷蛋白消化物培养基均可被检出。
新制备的培养基在2 天内不使用,最好置2 ~ 20℃暗处储存,可在2 ~ 3 周内使用。灭菌培养基保存在密封的容器中,可在半年内使用。如果指示剂颜色符合要求,或每3个月测试培养基的灵敏度应为合格。
3. 选择性培养基
(1 )对氨基苯甲酸培养基(用于磺胶类药物的无菌检查):照需气菌、厌气菌培养基及真菌培养基的处方及制法,各加对氨基苯甲酸0.125 g (培养基5),榕解后,摇匀,分装,灭菌。
(2)聚山梨酸培养基(用于油剂药品的无菌检查): 照上述需气菌、厌气菌培养基及真菌培养基的处方及制法,每1αm d 培养基中各加10 ml 聚山梨酸80 (培养基6),摇匀后,分装,灭菌。
4. 培养基测试
无菌检查用培养基、无论是市售的脱水培养基或配制的培养基,灭菌后,应澄清,无沉淀。此外,尚需进行无菌试验:将已备妥的培养基,按其用途分别置于30-35℃培养48 h, 20-25℃培养3 天均应无菌生长。如有菌生长须重新制备培养基,否则不能保证无菌检查结果的可靠性。BP 1998、USP24 均规定培养14 天不得有微生物生长,这一规定是合理的。
二、灵敏度试验
1. 菌种
各国药典对无菌检查用培养基的质量,高压蒸汽灭菌后,每一批均规定用已知不同菌株做生长试验来监控。根据加人定量菌的生长结果评价所用培养基的灵敏度是否符合规定,合格者方能作为无菌检查用。中国药典与英、美药典规定的菌种(如表1-4):需气菌有藤黄微球菌、金黄色葡萄球菌、枯草杆菌、铜绿假单胞菌;厌氧菌有生抱梭菌和普通拟杆菌;真菌有白色念珠菌和黑曲霉。加菌量皆在10- 100 个之间。
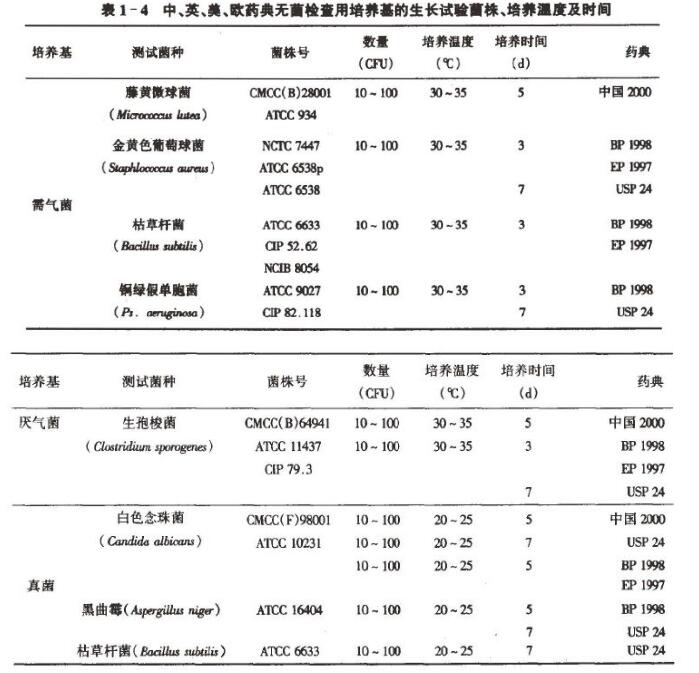
中国药典规定的培养基灵敏度试验菌株为藤黄微球菌[Micrococcus letea CMCC (B)28001 ]、生抱梭菌[ Clostridium sporogenes CMCC (B) 64941 ]和白色念珠菌[ Candida albicans CMCC ( F) 98001 ] 。藤黄微球菌是自然环境中常见的非致病菌,对营养要求较严格,生长较慢,作为需气菌的代表菌株较金黄色葡萄球菌、枯草杆菌具有更多的优点。BP 1998 增加铜绿假单胞菌、枯草杆菌作为培养基促生长试验的需气菌菌株,与前几版不同。值得关注的, USP24 ( 2000)无菌检查用需气菌、厌气菌培养基促生长试验所用菌株为金黄色葡萄球菌、铜绿假单胞菌、生抱梭菌;大豆酷蛋白消化物培养基促生长试验所用菌株为枯草杆菌、白色念球菌、黑曲霉菌,其中铜绿假单胞菌、黑曲霉菌为新增加的菌株。
2. 细菌计数方法
(1)细菌标准浓度比浊法:取藤黄徽球菌、白色念珠菌培养18 h 左右的琼脂斜面菌苔,刮少许加至适量无菌生理盐水或缓冲液管内混匀,制成均匀的菌细胞悬液;取生抱梭菌不含琼脂的需气菌、厌气菌培养基新鲜培养物,吸至灭菌离心管,离心,弃去上清液,菌体用0.9%无菌氯化纳溶液制成均匀菌悬液。分别取其1 ml 加在空标准管内,用中国药品生物制品检定所监制的细菌标准浓度比浊管比浊,与测试菌液管并列,在细菌比浊图片纸上对比观察各种线条的透黑度,两者一致时表示浓度相当。如待测菌液浓度大(即透黑度低于标准管浊度时),可加0.9% 无菌氯化钠溶液调节使之最接近,记下所用的无菌氯化纳溶液总量。然后根据标准比浊管所代表的各种细菌标准浓度(请看说明书)的每毫升含菌数计算,即可得知待测菌液的近似浓度,并将其用0.9%无菌氯化锅溶液或营养肉汤培养基稀释成每毫升含菌10 亿的菌液作为原液备用。临用前再将原液10 倍稀释至10-7,每毫升含菌大约100 个(包括死菌体在内) 。如欲知其所含活菌数,须按平板菌落计数法,取其1 ml 菌液加在溶化并冷至45℃的营养肉汤琼脂中混匀,倾注平皿, 培养,计数菌落数即可。按标准管比浊制成的10-7菌液,经验表明,活菌数一般每毫升均在50 ~ 100 个之间。用营养肉汤培养基稀释的标准菌液,于冰箱保存,1 周之内活菌数变化较小。
(2)原菌培养液直接稀释法:取已知菌的新鲜培养物少许,接种在相应的10 ml 液体培养基中,按规定的最适温度和时间培养后作为原菌液。然后取原液1 ml 作10 倍稀释至10-7 ,制成1 ml 含10- 100 个菌的稀释菌液(并用平板法计数)。在相同条件下,经多次测定证明后,稀释液可直接使用,不必每次测定活菌数。
将藤黄微球菌的上述(1)或(2)稀释菌液1ml,接种至每管装量为9 ml 的需气菌、厌气菌培养基3 管;生抱梭菌的上述(1)或(2)的稀释菌液1 ml,接种至每管装量为12 ml 的需气菌、厌气菌培养基3 管;白色念珠菌的上述(1)或(2)的稀释菌液ml,接种至每管装量为9 ml的真菌培养基3 管。以未接种的培养基作对照,按规定的温度培养5 天并逐日记录结果。
结果判定:以每株菌接种后的各培养基均不得少于2 管呈现生长,即该培养基的灵敏度检查符合要求。
(3 )培养基临用前的检查
需气菌、厌气菌培养基在临用前须做检查,培养基上部约1/10-1/5 处呈现淡红色时可以使用,若淡红色部分超过1/3 高度时,应将培养基用水浴或其他方法加热,直到无色后,冷却至45℃以下时再立即接种检品。但用沸水加热法去除培养基内游离氧时,每批培养基只限加热一次,否则影响培养基的质量。全管呈现淡红色时,不得再用。
全部 0条评论